Medical Devices that are being sold in Europe are required to be CE Marked under the Medical Devices Regulation (typically shortened to 'MDR'). Such devices placed on the market in Great Britain must, for the foreseeable future meet either the EU Regulation or the UK Medical Devices Regulations 2002
Devices that are intended to be used for medical purposes are likely to be required to be CE marked under the Medical Devices Regulation (EU 2017/745), however it is important to note that not everything found in a hospital or in a healthcare setting will fall within scope. Only devices or accessories of such devices that fulfil a specific definition can be CE marked.
The Regulation defines a medical device as any instrument, apparatus, appliance, software, material or other article that is intended to be used for the purposes of (where appropriate) diagnosis, prevention, monitoring, treatment, alleviation or compensation of a disease, or an injury or a handicap. The Directive also covers ‘devices’ used for the investigation, replacement or modification of the anatomy or of a physiological process, and of control of conception. As with the majority of the Directives, there is also a list of exclusions, the most prominent being the In-Vitro Diagnostic Medical Devices Regulation. A major change from the previous Directive is the amalgamation of Medical Devices with active Implantable Medical Devices which now fall within the same Regulation
Many of the other ‘Product Directives’ have links to the Medical Devices Regulation; predominately related to exclusions. For example, devices covered by the MDR, are excluded from the Low Voltage, ElectroMagnetic Compatibility, Pressure Equipment (Category 1 and SEP only) Directives. The MDD also states that were a relevant hazard exists, devices which are also machines as per the Machinery Directive, shall also comply with the relevant Essential Health & Safety Requirements of the Machinery Directive. The requirement shall only be applied to the extent where they are more specific than the essential requirements set out in the MDR.
The route to conformity is very much dependent upon the classification of the device. Class I devices can be self-declared, where any other class requirements the involvement of a Notified Body. Classification is decided upon establishing the applicable rule within Annex IX of the Medical Devices Directive. From here the route to conformity is explained within Article 11 and the applicable Annex.
For Class I devices (other than custom-made or devices intended for clinical investigations), the conformity assessment procedure is set out in Annex VII and is known as ‘EC Declaration of Conformity.’ The procedure is summarised below:
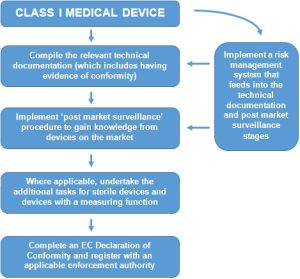
Whilst other Product Legislation requires manufacturers to undertake a risk assessment, the Medical Devices Directive goes further and effectively requires a Risk Management System (RMS) to be implemented. The RMS not only deals with the identification of the initial hazards associated with a device, but also has the procedures for undertaking post market surveillance. Whilst the Directive does not set out a formal procedure, a harmonised European Standard (EN ISO 14971) is available to be used.
Finally, the requirement to register the company with an enforcement authority is unique to the Medical Devices Directive. In the UK, manufacturers would register themselves with the MHRA (at the cost of a small administration charge). Registration is on a per-product-code basis, not necessarily on a per-product basis. Multiple codes may apply where a manufacturer has a wide product portfolio, however the registration form allows this to be done in a single application.
Please always refer to the Directive for the full text and definitions to help understand the application and requirements of the Medical Devices Directive
The CE Marking Association have a range of services to assist manufacturers, importers and users of Class I Medical Devices with a range of services, as well as providing advice for the Class IIa & b and Class III Devices too. Here are some of the services on offer:
For more support on CE Marking under the Medical Devices Directive, or to find out how we can help you, please contact us as below: